[2026-06-08] (Accès libre) En octobre 2024, nous avions publié un article intitulé « Une révision rapide du RDM et du RDMDIV : rêve ou réel espoir ? ». Plusieurs actualités de ces dernières semaines nous semblent mériter une mise en perspective.
Rappelons que les périodes transitoires pour les dispositifs hérités (« legacy » en anglais) doivent se terminer :
- le 31 décembre 2027 pour les dispositifs de classe III et pour les dispositifs de classe IIb implantables, sauf exception sutures, agrafes, etc.
- le 31 décembre 2028 pour les autres dispositifs médicaux.
Vous avez signé un accord écrit avec un organisme notifié avant le 26 septembre 2024 et bénéficiez de la période transitoire.
On parle de révision du RDM… Peut-être vous interrogez-vous sur l’opportunité de soumettre votre documentation technique rapidement, alors que des changements importants et favorables pour vous pourraient survenir ?
Des impératifs de sécurité
N’oublions pas que la réglementation vise d’abord à protéger les patients et les utilisateurs des dispositifs médicaux. Face aux risques posés par des dispositifs dangereux ou potentiellement dangereux, les autorités restent, bien sûr, toujours prudentes.
L‘ANSM a ainsi pris une décision le 10 avril 2026 quant au dispositif de stérilisation définitive FemBloc (qui n’est pas un dispositif hérité). Elle suspend la mise sur le marché et l’utilisation en France, excepté dans le cadre d’un essai clinique, de ce dispositif pourtant certifié par TÜV SÜD le 11 mars 2025. Cette décision peut surprendre, mais il y a eu un douloureux précédent, le dispositif de stérilisation définitive ESSURE, commercialisé en France entre 2002 et 2013, dont l’ANSM surveille toujours les effets.
Par ailleurs, l’ANSM a renforcé la surveillance des implants de renfort pariétal (indiqués dans le traitement des hernies). Les signalements d’effets indésirables par les patients ont en effet beaucoup augmenté en 2025 et 2026, alors que ces produits présentaient un profil de risque stable jusqu’en 2024.
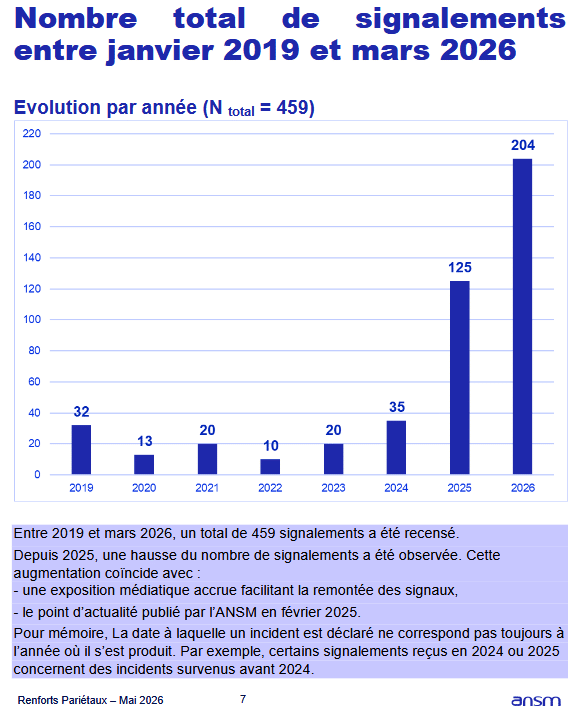
Les produits n’ont pas changé. Mais la remontée d’informations des patients, bienvenue et encouragée depuis plusieurs années, est là. Les attentes des patients et des professionnels de santé augmentent. Certains effets indésirables ne seront probablement plus considérés comme acceptables.
Par conséquent, pour certains produits, il pourrait devenir rapidement difficile de se réfugier derrière un discours du type « ces produits sont sur le marché depuis des années sans problème ».
Les autorités auraient du mal à accepter, et justifier vis-à-vis des patients, les risques posés par des dispositifs dont la conformité au RDM n’a pas été démontrée, alors que le choix d’approvisionnement en dispositifs alternatifs existe par ailleurs.
Une concurrence internationale dynamique
Le 8 mai 2026, la Commission européenne a publié des informations nouvelles et instructives sur la compétitivité des fabricants européens. La 19ᵉ mise à jour de l’enquête auprès des organismes notifiés sur la disponibilité des dispositifs médicaux (DM) présente en effet le nombre total et la distribution en taille des fabricants situés dans l’Union européenne (UE) et en dehors de l’UE. Il s’agit des entreprises clientes des organismes notifiés, donc des fabricants de dispositifs nécessitant une certification au titre du RDM (classes III, IIb, IIa, Im, Is, Ir).
Notons déjà un nombre de fabricants hors UE supérieur au nombre de fabricants dans l’UE. En outre, la répartition en taille des entreprises est défavorable à l’Union européenne. Le graphe dénombre 3128 entreprises de taille moyenne à grande hors UE, face à 1885 entreprises de mêmes catégories dans l’UE. À l’inverse, c’est dans l’UE qu’on trouve le plus de micro-entreprises.

Or, il y a bien une taille critique à atteindre pour pouvoir absorber les coûts de certification et autres coûts de mise en conformité et de maintien en conformité. Plus les entreprises sont grandes, plus elles sont susceptibles de répartir les coûts d’un certificat du système de management de la qualité sur un grand nombre de dispositifs. MedTech Europe l’a très bien démontré dans son rapport (page 42) intitulé « MedTech Europe IVDR & MDR Survey Results 2024 -Public Report December 2024 » (voir notre article).
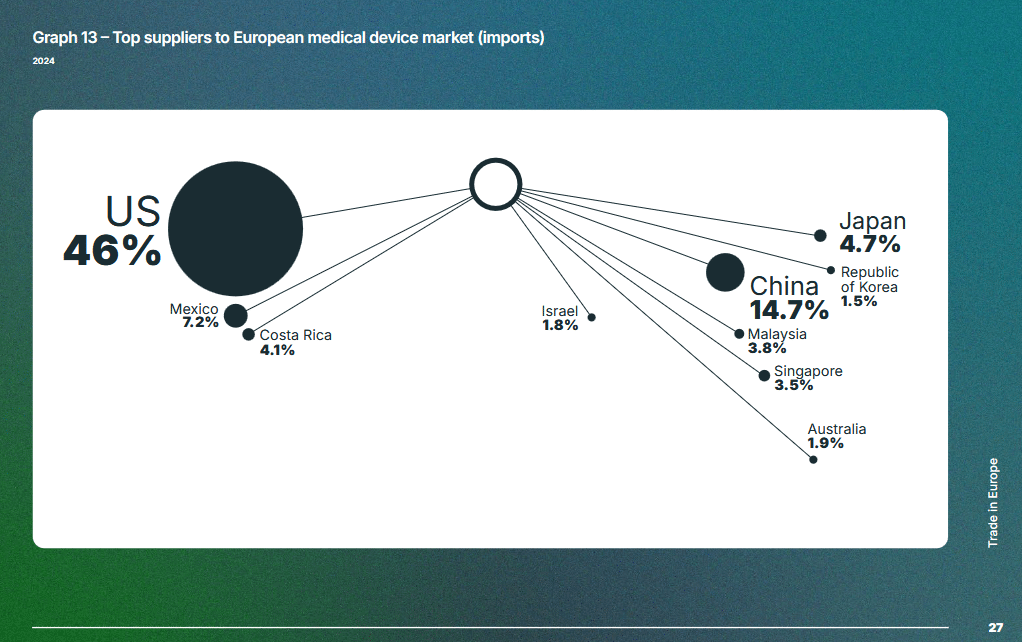
Le rapport annuel sur le marché des DM du syndicat MedTech Europe montre l’origine principale de ces entreprises : États-Unis, Chine, Mexique et Japon. Voir le graphe ci-dessous, toutes classes de dispositifs confondues et en incluant les DM de diagnostic in vitro. La diapositive 25 de ce rapport mentionne une balance commerciale encore positive en 2024, mais un afflux d’importations en provenance des marchés extra-européens.
Des mesures de protection des petites entreprises (européennes) incertaines
L’Union européenne dit vouloir protéger les intérêts des entreprises micro, petites ou de taille moyenne.
Ainsi, le projet de simplification du RDM prévoit à travers l’article 50 des réductions de tarifs de certification pour les petites entreprises. Il a conduit Team-NB, l’association européenne des organismes notifiés, à demander une expertise légale, publiée le 26 mai 2026. L’expertise conclut que le projet d’article 50 serait nul car il contreviendrait à la Charte des droits fondamentaux de l’UE.
Par ailleurs, la préférence européenne n’est pas la règle en matière d’achats. Le règlement d’exécution (UE) 2025/1197 excluant les entreprises chinoises de certains marchés publics de dispositifs médicaux de l’UE est une mesure exceptionnelle à ce jour (voir notre article). Certes, le 12 mai 2026, le projet d’acte législatif sur les médicaments critiques a obtenu un accord provisoire entre le Conseil et le Parlement (voir le communiqué de presse). Il pourrait constituer un précédent transposable aux dispositifs médicaux, mais cela prendra encore du temps.
Un goulet d’étranglement en perspective
Autres données nouvelles dans la 19ᵉ mise à jour de l’enquête auprès des organismes notifiés sur la disponibilité (par rapport à notre article du mois dernier qui analysait les données arrêtées au 31 octobre) : les reports de planning demandés par les fabricants (diapositives 30 et 31). Il s’agit de :
- reports de soumission de la documentation technique : 80 % des ON voient 25 à 75 % de leurs dossiers retardés, avec un impact sur la planification de leurs activités.
- reports d’audit du système de management de la qualité : moins fréquents, ils concernent tout de même 60 % des ON.
Le tableau de bord le plus récent de l’enquête est accessible ici. Comme le montre le graphe ci-après, le nombre de certificats émis au titre du règlement sur les DM (RDM, ou MDR en anglais) ne progresse donc pas rapidement. Il semblerait même reculer, mais rassurez-vous, cela s’explique par un changement dans le mode de calcul.

En outre, les organismes notifiés (ON) traversent une période difficile selon Team-NB, qui représente environ 80 % des ON et des certificats délivrés au titre du RDM. Son rapport annuel sur le secteur, en date du 18 mai, montre que les ON ont dû licencier du personnel technique en 2025 et indique que certains d’entre eux devront peut-être fermer. Le nombre de soumissions reste effectivement en deçà des prévisions et continue à diminuer. Team-NB s’attend à ce que cette tendance perdure jusqu’à l’issue du projet de simplification des règlements.
En cohérence avec les données de l’étude de disponibilité (92 % des organismes notifiés acceptent de nouveaux clients (diapositive 24)), Team-NB confirme que les ON ont actuellement la capacité de prendre des évaluations. Mais on peut s’attendre à un goulet d’étranglement à l’approche de la fin de la période de transition (fin 2027 pour les classe III et IIb implantables, fin 2028 pour les autres).
Par conséquent, si vous envisagez de repousser la date de soumission de votre documentation technique, vous risquez un refus de votre organisme notifié.
Les axes de simplification du RDM
Saluons déjà les progrès dans le domaine :
- de l’harmonisation des pratiques des organismes notifiés (voir notre article dans ce numéro sur le règlement d’exécution (UE) 2026/977, d’application obligatoire en février 2027), et
- des « technologies bien établies » (« well-established technologies », voir notre article sur les règlements délégués en cours d’examen final, entrée en vigueur potentielle en juillet 2026). Signalons ce 8 juin la déclaration conjointe de Team-NB, de sociétés d’assurance, et d’associations de médecins et d’organismes notifiés, qui pointent des catégories de dispositifs trop vagues et pour certains retirés du marché après des incidents graves.
En outre, le projet de simplification du RDM, publié le 16 décembre 2025, est toujours en consultation.
Attention ! La date limite de cette consultation semble fixée au 3 août 2026. Cependant, les équipes de la Commission se concentrent sur les sujets les plus brûlants et interrogent les parties prenantes pour identifier les priorités. Un premier jet de rapport est prévu dès la mi-juillet 2026 et le projet législatif serait soumis au Parlement européen début 2027.
Faites entendre votre voix par la consultation en ligne ou à travers vos députés ! Vous pouvez retrouver une synthèse et les liens vers les textes dans notre article de janvier. Voici quelques exemples d’information qu’il nous semble utile de remonter :
- L’évolution des coûts annuels de certification et des autres coûts ou équivalents temps plein, occasionnés par le passage de la directive au RDM, par exemple, en proportion du chiffre d’affaires et/ou de la masse salariale ;
- Vérifier dans quelle mesure l’application de la nouvelle annexe VII ((UE) 2026/977 cité plus haut) aurait amélioré ou pourrait améliorer le déroulement de votre projet de certification. Des mesures supplémentaires sont-elles nécessaires ?
- Êtes-vous dans une situation de niche ou mono-produit, qui limite votre capacité à absorber les coûts de certification ?
- Enfin, peut-être souhaitez-vous contester les exigences d’un organisme notifié ? Auriez-vous besoin d’un médiateur ? L’article 35, paragraphe 6 bis, de la révision du RDM prévoit : « Un fabricant ou un organisme notifié peut saisir l’autorité responsable des organismes notifiés, de manière dûment motivée, de tout litige non résolu découlant de l’application des exigences énoncées à l’annexe VII et de la participation d’un organisme notifié à l’évaluation de la conformité conformément à l’article 52 et aux annexes IX, X et XI. L’autorité examine la question et rend sa décision dans un délai de 90 jours. » Cette mesure serait-elle utile pour vous ? Si oui, affirmez-le !
Conclusion
En conclusion, les certifications marquent une pause en raison du retard des fabricants, entraînant une sous-charge chez les ON qui ont dû licencier. Tous les ON peuvent aujourd’hui prendre en charge des évaluations. Leur capacité réduite s’avèrera peut-être critique quand il faudra évaluer un grand nombre de dispositifs à l’approche de la fin des périodes transitoires. La concurrence extra-européenne est forte sans protection garantie à court terme, même pour les petites entreprises. Notre recommandation est de sécuriser le plus rapidement possible votre (ou vos) premier(s) certificat(s) RDM, sans attendre des mesures hypothétiques. En parallèle, faire entendre votre voix à travers des données très concrètes.
Article rédigé par Muriel GONIDEC, Présidente de DM Experts SAS