[2025-02-17] (Accès libre) Swissmedic, l’Institut suisse des produits thérapeutiques, a évalué la conformité aux exigences de surveillance après commercialisation de 30 dispositifs médicaux (DM) de classes IIa ou plus.
Un court compte rendu de trois pages, publié le 17 février 2025 et intitulé « Dispositifs médicaux – Évaluation par Swissmedic de la documentation sur la surveillance après commercialisation », synthétise les résultats. L’évaluation porte effectivement sur un échantillon de 30 DM relevant de l’ancien droit, sur la base des données de vigilance.
Au final, 20 DM sur 30 présentent des non-conformités dans la documentation sur la surveillance après commercialisation. Pas moins de 85 non-conformités ont été observées au total, en relation avec les exigences du plan de surveillance après commercialisation, du rapport de sécurité et du système de surveillance après commercialisation.
La figure ci-dessous donne, à titre d’exemple, la proportion de non-conformités en fonction des documents évalués :
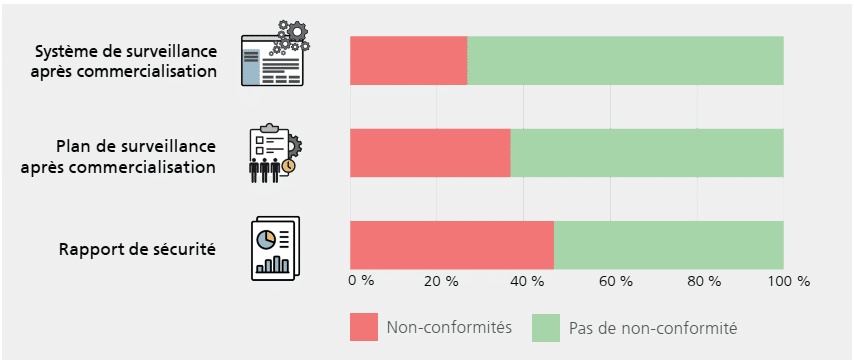
Pour plus d’informations, nous vous invitons à parcourir le document en détail.
L’étude porte évidemment sur les exigences réglementaires suisses de l’Ordonnance sur les dispositifs médicaux (ODim). Cependant, les articles de l’ODim se réfèrent très largement à la réglementation (UE) 2017/745, du moins pour le sujet présent. Les fabricants de l’Union européenne peuvent donc s’inspirer de ce retour d’expérience pour déceler par avance d’éventuelles non-conformités dans leur propre système.
Article rédigé par Christophe Saillet, membre du réseau DMEXPERTS.