[2026-02-02] (Accès libre) Depuis le 2 février 2026, le règlement sur le système qualité QSR (« Quality System Regulation ») est officiellement devenu le QMSR (« Quality Management System Regulation »).
A. Généralités
La FDA (Food and Drug Administration américaine) avait progressivement préparé la transition vers le QMSR en publiant notamment sur les derniers mois, une FAQ en août 2025, des présentations pédagogiques en septembre 2025, puis une proposition de guide en octobre 2025.
Dans ce contexte, un nouveau processus d’inspection est désormais appliqué, basé sur le manuel « Compliance Program Manual – 7382.850 » en remplacement du QSIT (« Quality System Inspection Technique » for device inspections).
Les programmes de conformité (« Compliance Programs ») de la FDA fournissent des instructions au personnel de la FDA afin de mener des activités visant à évaluer la conformité de l’industrie à la loi fédérale sur les aliments, les médicaments et les cosmétiques (« Federal Food, Drug, and Cosmetic Act » ou FD&C Act) et à d’autres lois administrées par la FDA.
Le document de 78 pages est intitulé « INSPECTION OF MEDICAL DEVICE MANUFACTURERS ». Il remplace le précédent manuel du même nom, publié le 29 septembre 2023 sous le numéro CP 7382.845, ainsi que le manuel de programme de conformité « Medical Device PMA Preapproval and PMA Postmarket Inspections – CP 7383.01 », publié le 5 mars 2012.
Ce nouveau manuel s’aligne selon les exigences du QMSR, lui-même aligné sur les exigences de la norme ISO 13485:2016 « Dispositifs médicaux – Systèmes de management de la qualité – Exigences à des fins réglementaires ». Il intègre ainsi les pratiques d’inspection basées sur le risque, actualise les références réglementaires, les procédures, et actualise les contacts de la FDA.
B. Contenu du manuel
Partie I : Contexte
La 1ère partie est consacrée à un rappel historique et aux liens avec la réglementation américaine existante, l’ISO 13485:2016, la Clause 3 de l’ISO 9000:2015, ainsi que les exigences supplémentaires de la FDA (voir OAFRs plus bas), de même que le lien avec le MDSAP (« Medical Device Single Audit Program »).
Sans surprise, les inspections menées auprès des fabricants ont pour objectif d’évaluer la capacité de leur système de management de la qualité (SMQ) à satisfaire aux exigences de la FDA, à fournir une assurance raisonnable quant à la sûreté et à l’efficacité des dispositifs médicaux (DM), et à vérifier que la gestion des risques et la prise de décision fondée sur les risques sont effectivement mises en œuvre au sein du SMQ.
Le manuel se focalise régulièrement sur les inspections liées aux soumissions de type « Premarket Approval » (PMA) avant et après commercialisation, points issus du manuel de programme de conformité CP 7383.01, cité précédemment.
Des instructions concernant la préparation et l’organisation des inspections sont ensuite données aux inspecteurs de l’« Office of Inspections and Investigations » (OII) dans les sections suivantes du manuel.
Partie II : La mise en œuvre
Cette partie explique :
- La priorisation et la planification des inspections : le programme d’inspections doit prévoir en priorité les fabricants « à risque » : fabricant de classe III n’ayant jamais été inspecté, fabricant de dispositifs implantables, de maintien de la vie, ou ayant une fréquence élevée de rappels, fabricant de nouveaux dispositifs sur le marché… ainsi que les inspections après commercialisation.
- Coordination : la nécessité de coordination entre l’OII et les autres centres dans le cas de dispositifs incluant des produits biologiques ou des médicaments, ou de dispositifs émettant des radiations, et dans le cadre du MDSAP.
Partie III : L’inspection
Cette partie centrale décrit la partie opérationnelle de l’inspection :
- Les inspections reposent sur une approche fondée sur les risques et s’organisent autour de 6 thématiques/zones (« Area ») selon le QMSR, complétées des 4 autres exigences supplémentaires applicables de la FDA (« Other Applicable FDA Requirements ou OAFRs »). Le processus d’inspection est illustré dans le manuel sous la forme d’un diagramme ; un tableau récapitulatif qui détaille le périmètre de chaque thématique ainsi que les critères d’exigences associés est disponible en Annexe A du manuel.
- les 4 OAFRs supplémentaires sont les suivantes :
- Medical Device Reporting (MDR) – 21 CFR Part 803,
- Reports of Corrections and Removals – 21 CFR Part 806,
- Medical Device Tracking Requirements – 21 CFR Part 821,
- Unique Device Identification (UDI) – 21 CFR Part 830.
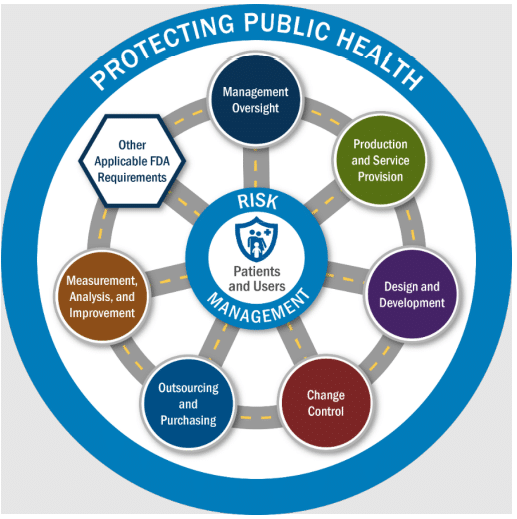
Diagramme 1 (issu du manuel) : « FDA Medical Device Risk-Based Inspections«
- De manière classique, afin d’identifier les risques et de définir les priorités d’inspection, l’OII analyse, en amont de l’inspection puis tout au long de celle-ci, un ensemble de données et d’enregistrements permettant d’identifier les risques associés aux produits et aux activités du fabricant : données de vigilance, rappel, réclamations, activités de surveillance après commercialisation, opportunités d’amélioration, etc.
- Le manuel décrit également les différents types d’inspection ainsi que le modèle d’inspection applicable selon le type d’inspection menée. Deux modèles d’inspection y sont présentés.
Quelques points d’instructions spécifiques abordés dans cette partie :
- validations des procédés : au moment de l’inspection, le fabricant devra avoir validé tous les procédés nécessitant une validation.
- conception et développement (C&D) : les fabricants doivent appliquer les exigences de C&D selon la réglementation FDA (§ 820.10(c), en lien avec la norme ISO 13485, clause 7.3. Ces exigences s’appliquent à tous les dispositifs médicaux, à l’exception de certains dispositifs de classe I ; seuls les dispositifs de classe I explicitement mentionnés dans le manuel sont concernés. Lorsque les activités de C&D sont confiées à un sous-traitant, le fabricant reste responsable et doit conserver ou avoir un accès aux dossiers de C&D, ainsi qu’à l’ensemble des procédures, documents et enregistrements associés aux dispositifs PMA en production.
- cybersécurité : pour les dispositifs « cyber » définis selon la section 524(c) du FD&C Act, leur conformité est également évaluée dans le cadre de l’inspection, le cas échéant. Le manuel renvoie à une FAQ publiée par la FDA sur ce sujet, que nous avions signalée dans un article précédent.
- vérification de l’enregistrement du fabricant et de la liste des dispositifs.
- ….
La section se termine par les attentes en matière de « reporting » des inspections.
Nous n’avons présenté qu’une partie de cette section, mais il s’agit sans doute de la section la plus intéressante avec l’annexe A pour un fabricant, car elle reflète directement les points d’attention des inspecteurs de la FDA dans le contexte et la mise à jour selon le QMSR.
Partie IV : Analytique
Cette partie décrit comment la FDA traite l’analyse physique ou chimique d’échantillons prélevés lors d’une inspection avec l’identification des laboratoires pouvant analyser les échantillons, les caractéristiques/types d’analyses possibles, ainsi que les méthodes d’analyse associées.
Partie V : Stratégie réglementaire/administrative
Cette partie explique comment la FDA conclut, suit et traite les résultats d’inspection :
- observations et constatations : celles-ci sont établies selon la gravité et la fréquence des défaillances. Des exemples sont fournis selon la situation ;
- décision réglementaire, classification finale de l’inspection : la FDA statue sur l’état de conformité globale du fabricant inspecté et indique le niveau d’actions attendues ;
- suivi et gestion administrative : les corrections et les propositions ou plans d’actions correctives, y compris les preuves des corrections déjà mises en œuvre, doivent être transmis par écrit par le fabricant dans un délai de 15 jours ouvrables après la clôture de l’inspection. Le manuel rappelle qu’une approche basée sur le risque doit être adoptée et appropriée afin de protéger au mieux la santé publique ;
- …
Partie VI : Références et contacts du programme
Cette partie liste :
- les documents de référence et sources de la FDA pouvant être consultés concernant la stérilisation,
- les contacts du programme (courriels, sites web,…) ainsi que les acronymes utilisés.
Annexes
Le document se conclut enfin par 2 annexes :
- Annexe A : Tableaux des « Areas » d’un SMQ, OAFRs, éléments et exigences.
- Annexe B : Évaluations réglementaires à distance (« Remote Regulatory Assessments » ou RRA), ces évaluations sont utilisées en remplacement ou en amont d’inspections sur site et utilisées dans certaines circonstances.
C. Conclusion
Ce manuel constitue la référence opérationnelle pour comprendre la manière dont la FDA inspecte les fabricants de DM dans le cadre du QMSR. Une lecture attentive et approfondie de ce document est fortement recommandée pour les fabricants concernés par la mise sur le marché aux États-Unis.
Un fabricant dont le système qualité est conforme au RDM (UE) 2017/745 et à l’ISO 13485:2016 est déjà bien positionné pour répondre aux exigences du QMSR, mais il ne doit pas oublier de prendre en compte les exigences particulières de la FDA qu’il faudra intégrer et documenter.
Article rédigé par Lam-Xé NOEL, Directrice Générale de DM Experts SAS